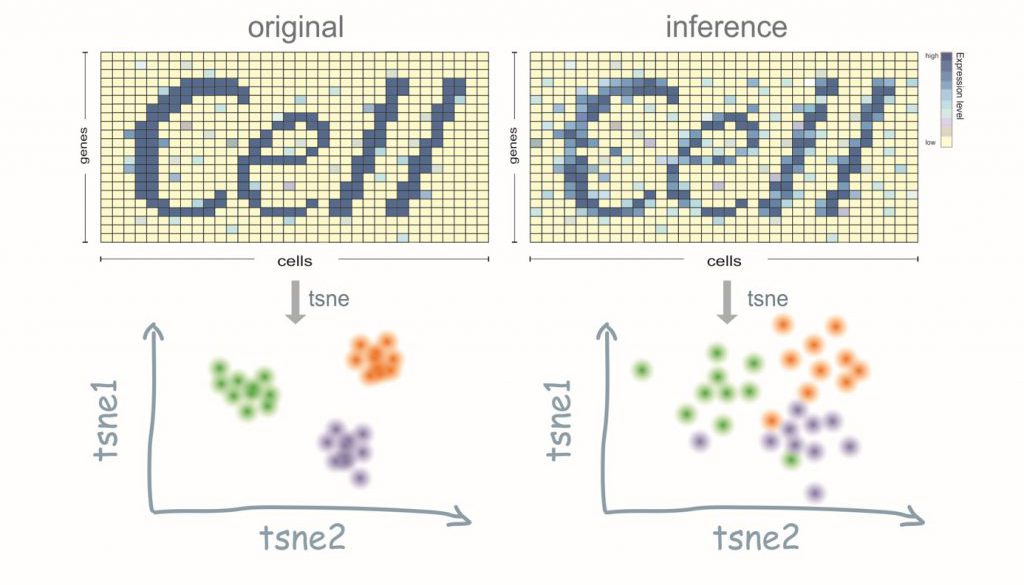
Pooling strategy on single-cell sequencing
Single cells, as the basic components of life, are a new window to understand individual differences among cells. With the development of advanced technologies to capture single cells quickly and accurately, scientists can narrow down their view from the bulk sequencing of thousands of cells, which averages out the cellular difference, to the subtle changes between individual cells. The elaborate atlas of single cell has shed light on multiple biological questions like revealing new cell types in cancers, investigating the dynamics of developmental processes, linkage and developmental trajectory of immune cells in cancer and identification of gene regulatory mechanisms.
The tantalizing possibility of figuring out the mechanisms of a lot of diseases, as well as identifying cells with a much clearer picture spawned dozens of papers and projects exploring the filed. Broad Institute together with many other organizations launched Human Cell Atlas Project . Allen Institute initialized the Human brain atlas etc.
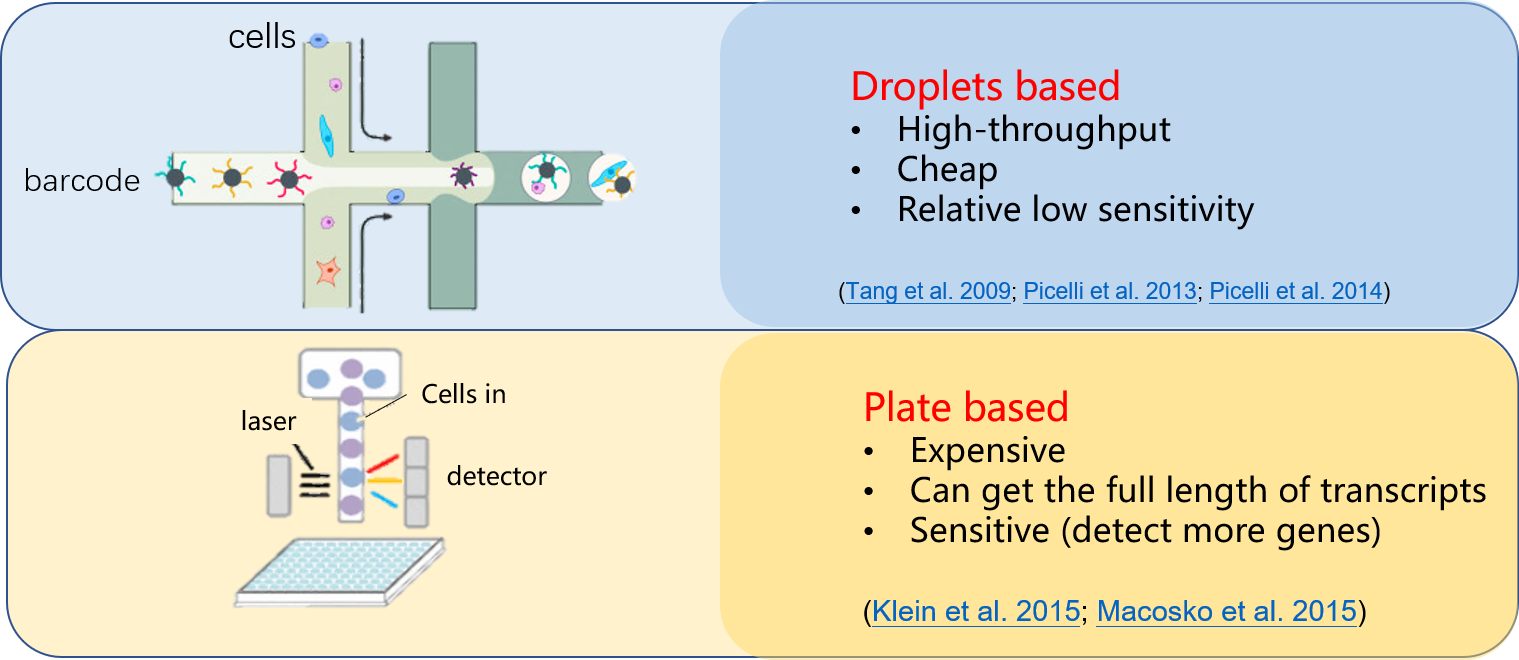
Folks not working in this field may curious about what technologies can reveal the single cell scale. Below I used a very simple illustration to show the most widely used two strategies for single-cell sequencing ( by saying sequencing, here we mainly focused on sequencing the transcriptomes.). The video produced from Harvard University introduced the droplet technology invented by Dave Weitz group ( interestingly, Dave is also the guest professor at my home university and I met him first introduced this idea in 2016 in China).
Well, now you had a basic idea on how to capture single cell from organisms, at least you believe there is a way we could extract the inner information of the cell.
The next question is, what will these data look like compared to the previous method like bulk RNA-seq (which means you could acquire the information of a sample with a combination information come from all cells in this tissue or the sample you feed into the machine. An obvious observation would be that a single cell actually carries much less nucleus than a bunch of cells, which to say, the possibility to detect a full range of genes is impossible. On the one hand, the feed-in material is little (like nucleus), on the other hand, there is a large variance among different cells (both cell types and cell fates, phases, environment etc). In a word, the expression profile for a single cell would be very sparse!
Now, since we’ve already known that the output of expression profile ( A matrix, where each row is a gene and each column represents a cell. If A_ij = 0, that indicates we didn’t detect gene i in cell j) is sparse in the context of single cell, but what could this property help us? If we looked into the technologies that we talked earlier, they seems to evolve into two directions:
- Cheap but fewer genes to be detected or Expensive but sensitive.
The main cost for the plate based method actually comes from library construction, which is a neccessity before you could let the sequencer read your sequence. Now, we imagine that, the sparsity could play a role to close this gap! – between cost and sensitivity. The scheme of the idea was presented below.
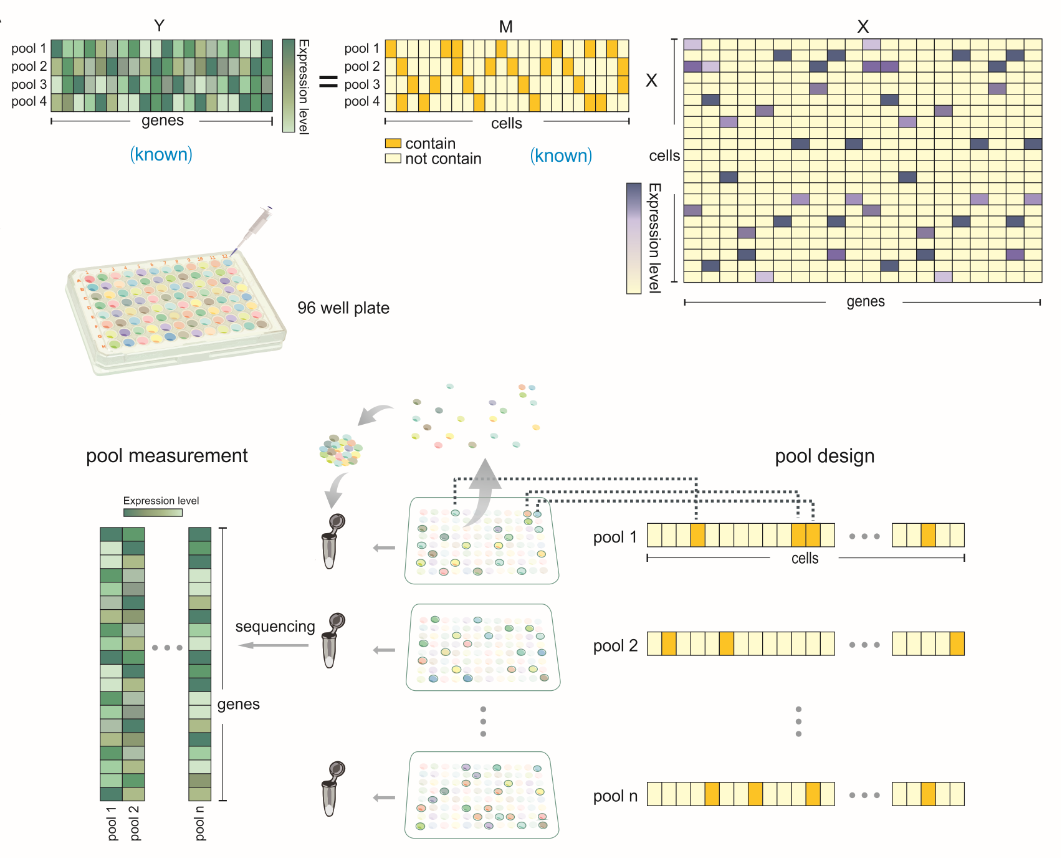
Rather than build a library for every single cell, we divided all cells into overlapped groups. Here overlapped means one single cell can appear in different pools, and one pool contains a group of cells. This overlapping fashion utilizes the same cell as an information bridge and can reduce the tests by cross information. Now, we could build and sequence to each pool rather than each cell and then decode each cell’s information according to this combination.
The sparsity of single-cell expression profile which suggests it is compressible. Compressed sensing is a widely used method for sparse signal recovery and sampling. It is an unbiased method and can guarantee to recover the exact signal if we had enough sample times according to circumstances. It has so many applications which most of them take advantage of the sparsity of the signal. For example, MRI, in order to get MRI picture people do not detect 360 degrees to form a 2D picture. Instead, the machine only does limited sampling for each section of the body since the MRI 2D figure will be a sparse signal after using some non-linear transformation into some other space. In this way, both the cost and the influence on the human body were decreased to a large extent.
The result of this method actually works out fine. In our test, instead of using more than five thousand libraries, just 1200 are enough for us to capture the cell’s identities (in another word, classification).
However, in this scenario, we also suffer from many constraints such as:
- The compress sensing method can only return sparse results, even though it’s very accurate we may still lose some information.
- If we only want some classification, the low sensitivity method like 10X, can also achieve that even with lower cost.
However, the bottom line is that we want to remind people in the field to appreciate more of their data’s intrinsic features and took advantage of that, maybe we could get very interesting results!